Scurfy mice harbor natural mutation in FOXP3 gene that clinically resembles FOXP3 deficiency. Both scurfy mice and genetically modified FOXP3-KO mice die prematurely within first month of life. In humans, clinically observed FOXP3 deficiencies do not seem to be as lethal as in mice though one could argue that scurfy mice lifespan could be extended if these mice were given human-like medical attention (for example, i.v. feeding, anti-inflammatory medication, so on).
In this regard, a new paper in Journal of Experimental Medicine is of great interest. It showed that a member of gut microflora, Lactobacillus reuteri, a probiotic microbe, when given orally could drastically extend lifespan of scurfy mice (30d vs. >125d) via microbiota–inosine–A2A receptor axis.


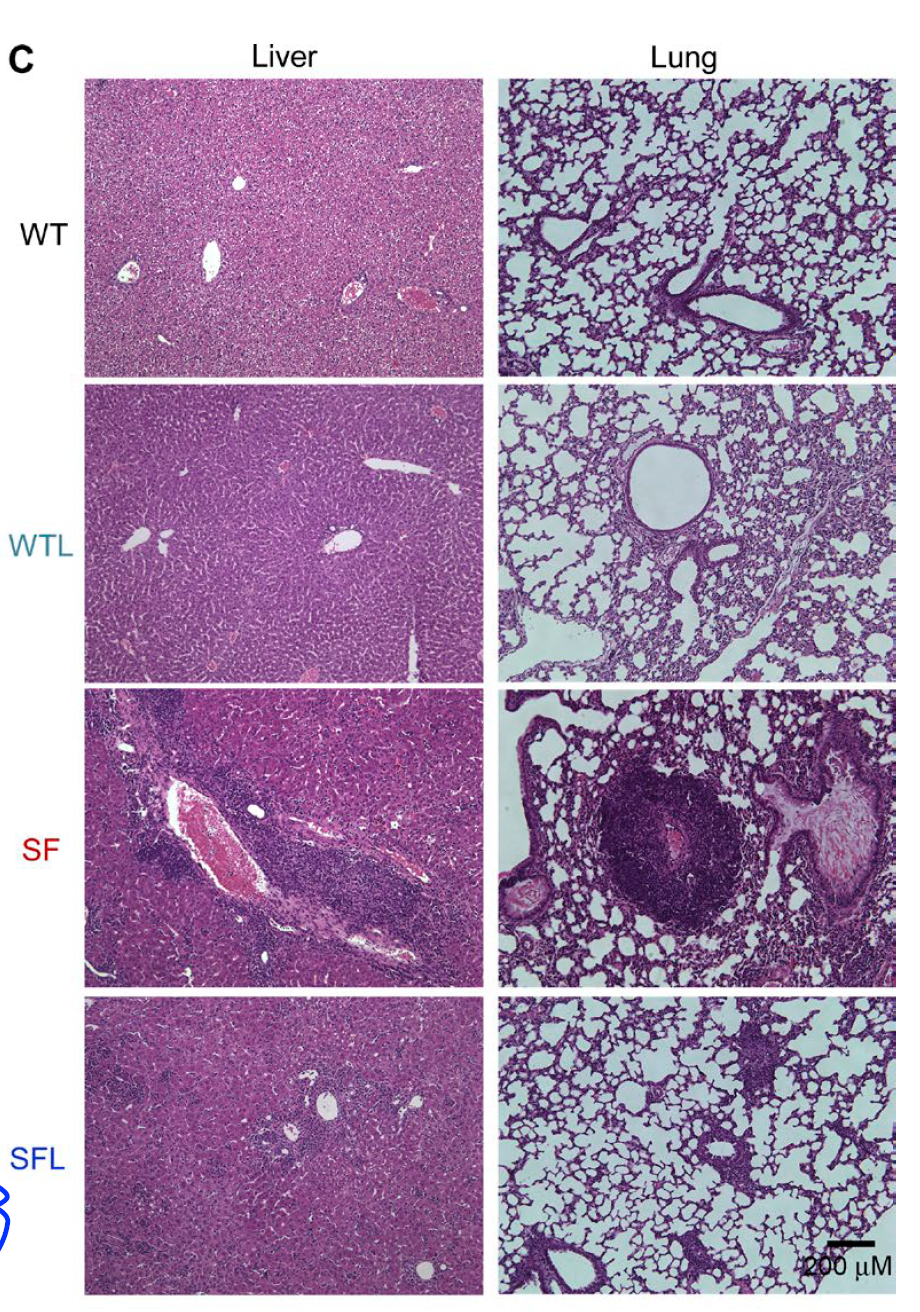
Morphologically, oral Lactobacillus reuteri significantly reduced tissue inflammation in scurfy mice.

Blood test showed that one molecule Lactobacillus reuteri could restore to WT level in scurfy mice was a purine metabolite inosine.

Indeed, oral inosine was able to recapitulate Lactobacillus reuteri effect on scurfy mice lifespan (and both Lactobacillus reuteri and inosine effects were specifically mediated via adenosine A2A receptor).


In summary, this study revealed that purine metabolites, inosine or adenosine could protect scurfy mice from tissue immunopathology and drastically prolong their lifespan. It is quite rare to see that one molecule could produce such effect. It would be interesting to see how caffeine consumption affects immunopathologies in humans as it acts as a natural antagonist to adenosine A2A receptor.
David Usharauli



