Immune system supposed to defend the body from infectious agents and genetically transformed cells (tumors). Usually, at the end of each immune response, that can be quite damaging (immune phase), immune cells will be involved in tissue healing, regeneration or remodeling (adaptation phase). However, sometimes infectious agents or tumors will circumvent these steps and jump directly to adaptation phase and will recruit immune cells to carry out their "agenda" at the expense of the host.
For example, we can speculate that tumor's "agenda" would be to grow and expand (metastasize). However, neither of these possible without local and distant tissue remodeling and its readiness to accommodate (accept) tumor cells. Here is where local immune cells become involved.
New study in journal Nature points to one of those possibilities. The authors showed that in a mouse model of mammary tumor metastasis, neutrophils promotes tumor lung metastasis via IL-17 produced by circulating γδ-T cells.
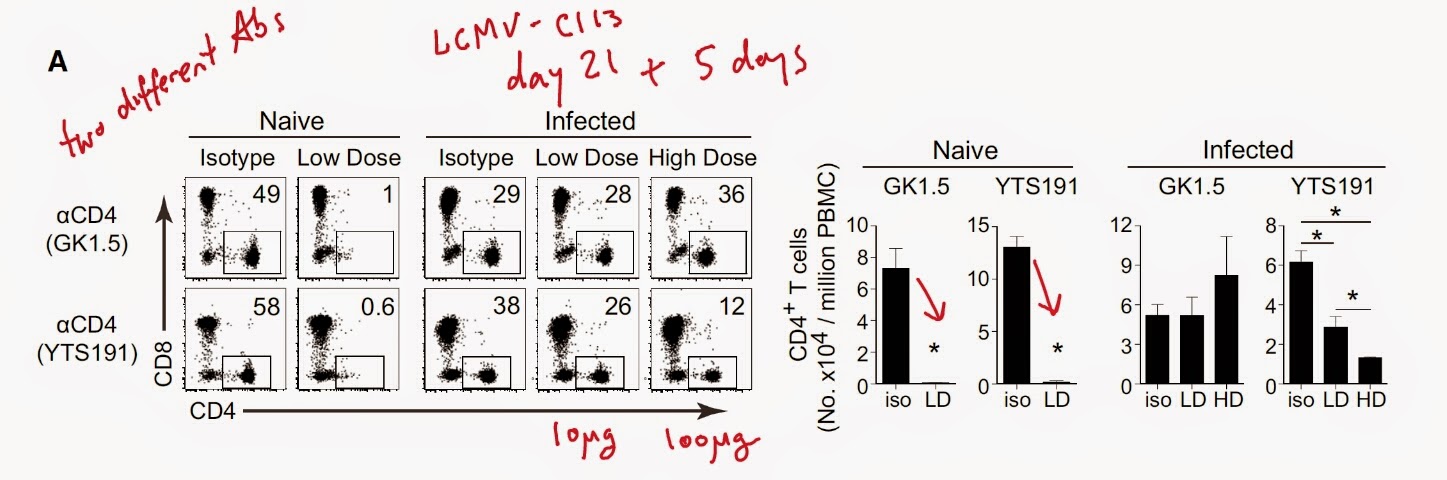
I would like to point out that the authors have used tumor transplantation model (to have shorter experiments) that is obviously very different from spontaneously arising tumors. Nevertheless, they found that lung or lymph node metastasis of skin transplanted tumor was reduced with neutrophil depletion using α-Ly6G antibody (this antibody supposedly selectively depletes neutrophils since they express it at high level).

Interestingly, tumor metastasis were also reduced in the recipients devoid of adaptive immune system and correlated with reduction of IL-17 and G-CSF (granulocyte colony-stimulating factor) serum levels.

Finally, the authors showed that γδ-T cell depletion or genetic deficiency reduced lung and lymph node metastasis of transplanted tumor.

In summary, these results suggest that tumor cells exploit not yet identified pathways within immune system (γδ-T cells / IL-17 / neutrophils axis) to prepare distant tissues to accommodate tumor colonies coming from original tumor niche. Only by understanding how immune cells interact with normal tissues during or after immune response, could we design ways to block such metastasis.
In general this paper is OK, especially if one considers other papers (two recent papers in JEM) corroborating the idea of IL-17's involvement in tumor initiation and metastasis. However, some data are not clear or not well explained, so not a Nature caliber paper, in my view. For example, in Fig. 3a, treatment with α-IL-17A did not modify IL-17 level in the serum. Also, the authors did not explain why they thought CD8 T cells were protective against metastasis in this model when tumor-bearing RAG KO hosts, which lack CD8 T cells, did not show increased metastasis (Fig. 2d versus. Fig. 3g)?
David Usharauli
I would like to point out that the authors have used tumor transplantation model (to have shorter experiments) that is obviously very different from spontaneously arising tumors. Nevertheless, they found that lung or lymph node metastasis of skin transplanted tumor was reduced with neutrophil depletion using α-Ly6G antibody (this antibody supposedly selectively depletes neutrophils since they express it at high level).
Interestingly, tumor metastasis were also reduced in the recipients devoid of adaptive immune system and correlated with reduction of IL-17 and G-CSF (granulocyte colony-stimulating factor) serum levels.
Finally, the authors showed that γδ-T cell depletion or genetic deficiency reduced lung and lymph node metastasis of transplanted tumor.
In summary, these results suggest that tumor cells exploit not yet identified pathways within immune system (γδ-T cells / IL-17 / neutrophils axis) to prepare distant tissues to accommodate tumor colonies coming from original tumor niche. Only by understanding how immune cells interact with normal tissues during or after immune response, could we design ways to block such metastasis.
In general this paper is OK, especially if one considers other papers (two recent papers in JEM) corroborating the idea of IL-17's involvement in tumor initiation and metastasis. However, some data are not clear or not well explained, so not a Nature caliber paper, in my view. For example, in Fig. 3a, treatment with α-IL-17A did not modify IL-17 level in the serum. Also, the authors did not explain why they thought CD8 T cells were protective against metastasis in this model when tumor-bearing RAG KO hosts, which lack CD8 T cells, did not show increased metastasis (Fig. 2d versus. Fig. 3g)?
David Usharauli























.png)
.png)