Here is a paradox for you that many don't even know that it exists: frequently, genetic immunodeficiency (weak immune response) syndromes are associated with immunopathologies (excessive immune response). So how could it be explained?
New study published in Journal of Experimental Medicine may have some new answers. This study described mouse model of human immunodeficiency syndrome called, Omenn syndrome (after its discoverer) and showed that immunopathology was driven by gut microbiota.
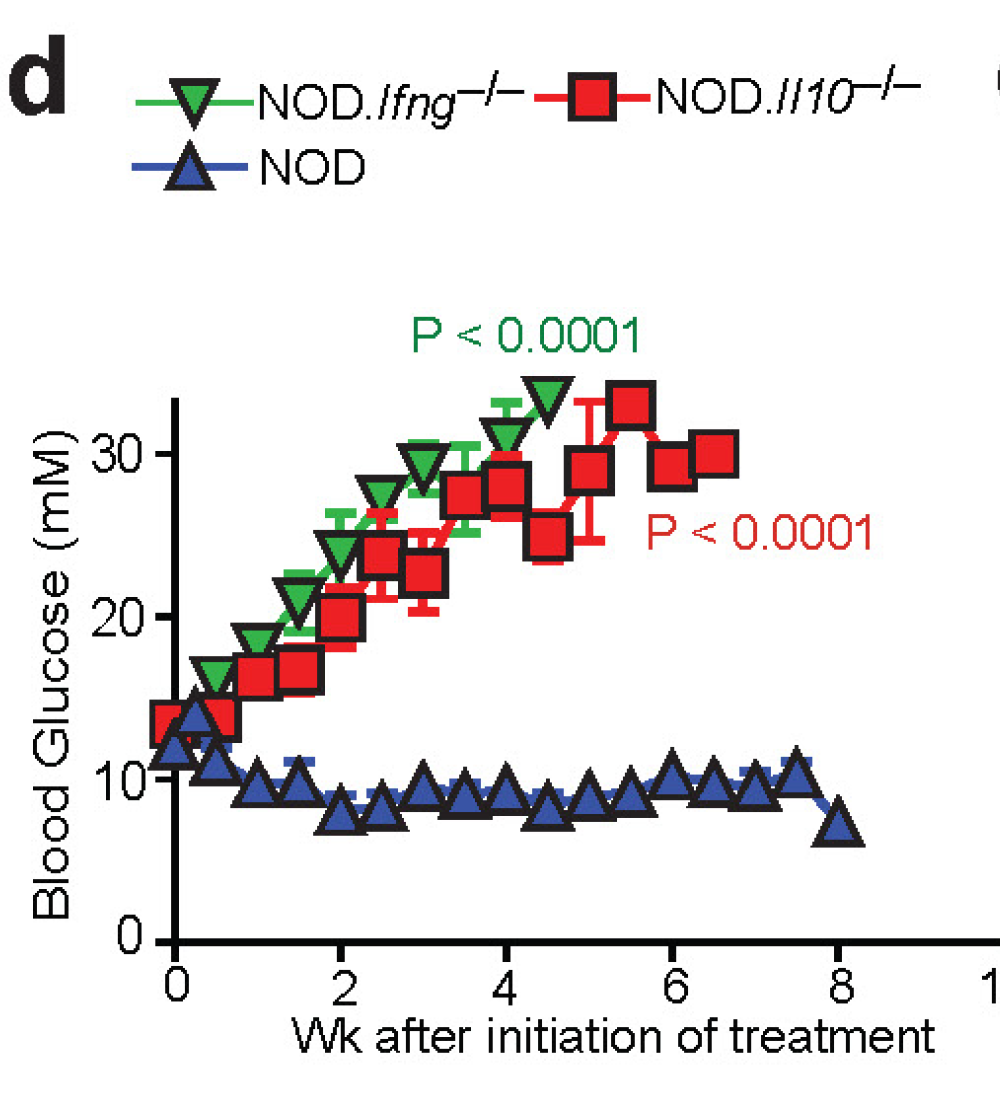
Omenn syndrome is caused by hypomorphic (low active) RAG mutations. Analysis of intestinal tissue from Rag2R229Q [Omenn] mice revealed pathological infiltration with inflammatory T cell subsets, TH17 and TH1.

Adoptive transfer showed that intestinal immunopathology was mediated by Rag2R229Q mutant T cells.

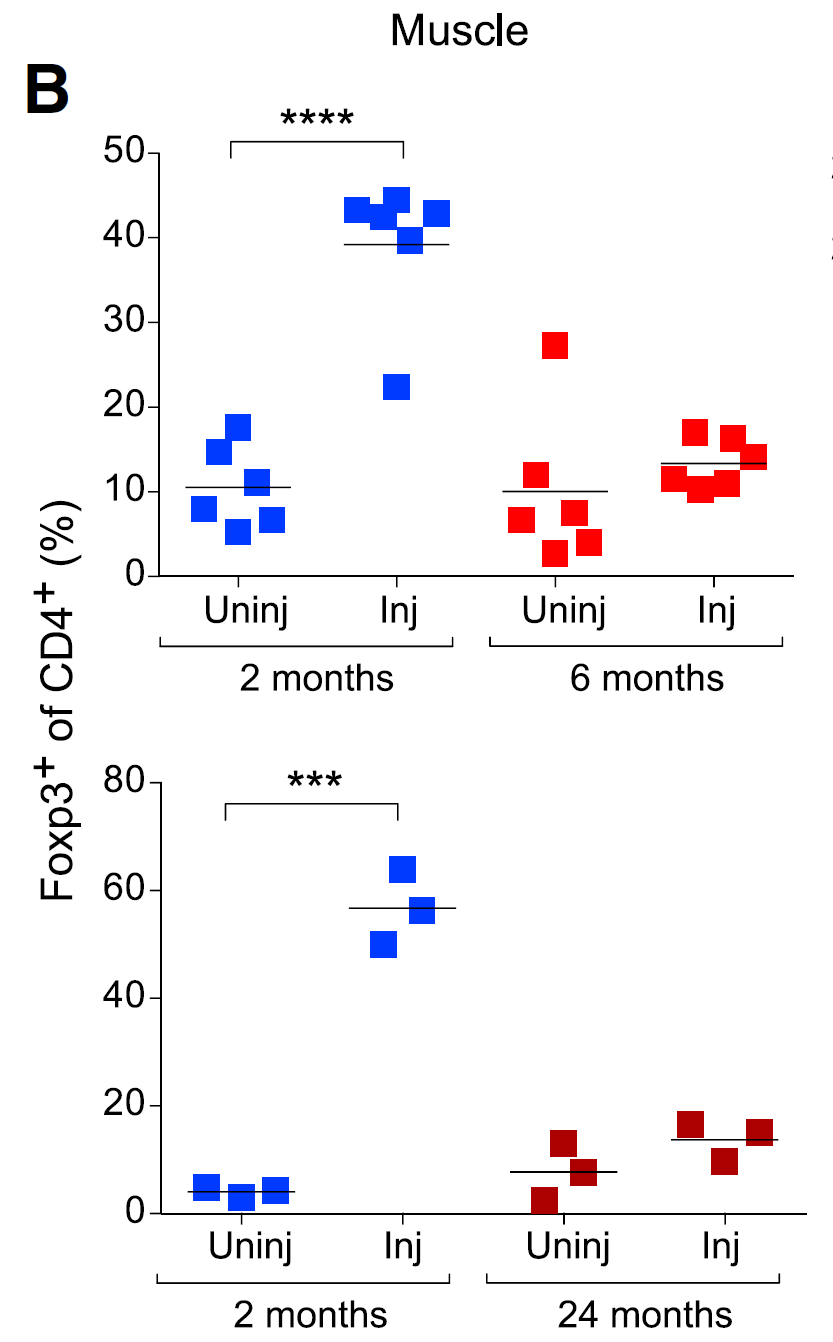
Interestingly, Rag2R229Q mouse harbored comparable numbers of Foxp3+ CD4+ T cells, implying that simple presence of regulatory T cells [generated in Rag2R229Q mice] was not enough to control tissue inflammation.

Defect of Tregs derived from Rag2R229Q mice was confirmed in adoptive transfer experiment with WT Tregs.

Besides T cell-driven immunopathology, Rag2R229Q mice displayed IgA deficiency (failure to properly coat gut microflora). These data pointed to the possibility of microbial translocation causing excessive inflammatory response.

Indeed, antibiotic treatment of Rag2R229Q mice could reduce intestinal immunopathology.

The role of antibiotic-sensitive gut flora in driving immunopathology in Rag2R229Q mice was confirmed in adoptive fecal transfer experiments.

In summary, this study suggests the following scenario: hypomorphic RAG defect in Rag2R229Q mice leads to "narrowing" of TCR and BCR repertoire. This in turn leads to outgrowth of oligoclonal T and B cells in Rag2R229Q mice (wherein Rag2R229Q mice contain T and B cells with limited, restricted, deficient TCR and BCR repertoires). Without proper TCR and BCR repertoire diversity, however, Rag2R229Q mice fails to develop tolerance (IgA and Tregs) to gut flora or commensal microbial antigens present at mucosal surfaces (such as lung, intestine). Repertoire restriction also leads to failure to mount adequate and proper immune response.
David Usharauli
Omenn syndrome is caused by hypomorphic (low active) RAG mutations. Analysis of intestinal tissue from Rag2R229Q [Omenn] mice revealed pathological infiltration with inflammatory T cell subsets, TH17 and TH1.

Adoptive transfer showed that intestinal immunopathology was mediated by Rag2R229Q mutant T cells.

Interestingly, Rag2R229Q mouse harbored comparable numbers of Foxp3+ CD4+ T cells, implying that simple presence of regulatory T cells [generated in Rag2R229Q mice] was not enough to control tissue inflammation.

Defect of Tregs derived from Rag2R229Q mice was confirmed in adoptive transfer experiment with WT Tregs.

Besides T cell-driven immunopathology, Rag2R229Q mice displayed IgA deficiency (failure to properly coat gut microflora). These data pointed to the possibility of microbial translocation causing excessive inflammatory response.

Indeed, antibiotic treatment of Rag2R229Q mice could reduce intestinal immunopathology.

The role of antibiotic-sensitive gut flora in driving immunopathology in Rag2R229Q mice was confirmed in adoptive fecal transfer experiments.

In summary, this study suggests the following scenario: hypomorphic RAG defect in Rag2R229Q mice leads to "narrowing" of TCR and BCR repertoire. This in turn leads to outgrowth of oligoclonal T and B cells in Rag2R229Q mice (wherein Rag2R229Q mice contain T and B cells with limited, restricted, deficient TCR and BCR repertoires). Without proper TCR and BCR repertoire diversity, however, Rag2R229Q mice fails to develop tolerance (IgA and Tregs) to gut flora or commensal microbial antigens present at mucosal surfaces (such as lung, intestine). Repertoire restriction also leads to failure to mount adequate and proper immune response.
David Usharauli